|
|
|
Widely distribution of hematological parameters in thalassemia patients with similar α-globin genotype |
Bijan Keikhaei1, Pejman Salehi-Fard1, Mostafa Paridar2, Mehraneh Karimzadeh3, Razie Dehghani4, Asma Zamiri5, Vahideh Takhviji6( ) ) |
1. Research Center for Thalassemia and Hemoglobinopathy, Health Institute, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran
2. Deputy of Management and Resources Development, Ministry of Health and Medical Education, Tehran, Iran
3. School of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran
4. Pediatric Department, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran
5. School of Medicine, Gorgan University of Medical Sciences, Gorgan, Iran
6. Laboratory Hematology and Blood Banking, School of Allied Medical Science, Shahid Beheshti University of Medical Science, Tehran, Iran |
|
|
|
|
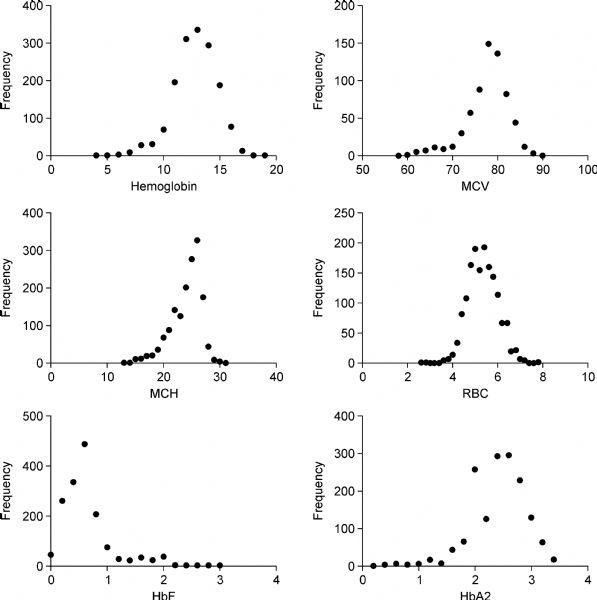
Abstract BACKGROUND: Thalassemia is known as the commonest monogenic disorder with an imbalanced rate of globin chains production of adult hemoglobin. Despite the available information about the thalassemia etiology, its phenotype varies from each patient to another. This study aimed to evaluate the hematological parameters of patients with the same -α3.7 homozygote and heterozygote genotypes to amend screening programs. METHODS: In this observational study, we evaluated 1301 thalassemia suspected patients who referred to the Thalassemia and Hemoglobinopathy Research Center of Ahvaz University of Medical Sciences, Khuzestan, Iran during 2014-2016. According to the genotyping studies, patients divided into 2 groups with -α3.7/aa (n = 646) and -α3.7/-α3.7 (n = 181) genotypes. Thereafter, distribution of hematological parameters evaluated in both groups. RESULTS: The mean age in heterozygous and homozygous groups was 25.7±4.5 and 26±4.4 years old, respectively. The degree of anemia was considerably varied in patients with the same genotype. MCV, RBC and MCH showed a wide distribution in patients. CONCLUSION: The findings presented here suggest that other molecular mechanisms along with α-globin gene mutations could be involved in determining the phenotypes of alpha thalassemia patients.
|
| Keywords
hematological parameters
α-globin genotype
alpha thalassemia
|
|
Corresponding Author(s):
Vahideh Takhviji
|
|
Online First Date: 23 October 2018
Issue Date: 30 November 2018
|
|
| 1 |
Alibakhshi R, Mehrabi M, Omidniakan L, Shafieenia S (2015). The spectrum of a-thalassemia mutations in Kermanshah Province, West Iran. Hemoglobin, 39(6): 403–406
https://doi.org/10.3109/03630269.2015.1070732
pmid: 26287614
|
| 2 |
Cao A, Kan Y W (2013). The prevention of thalassemia. Cold Spring Harb Perspect Med, 3(2): a011775
https://doi.org/10.1101/cshperspect.a011775
pmid: 23378598
|
| 3 |
Coelho A, Picanço I, Seuanes F, Seixas M T, Faustino P (2010). Novel large deletions in the human a-globin gene cluster: Clarifying the HS-40 long-range regulatory role in the native chromosome environment. Blood Cells Mol Dis, 45(2): 147–153
https://doi.org/10.1016/j.bcmd.2010.05.010
pmid: 20580289
|
| 4 |
De Gobbi M, Viprakasit V, Hughes J R, Fisher C, Buckle V J, Ayyub H, Gibbons R J, Vernimmen D, Yoshinaga Y, de Jong P, Cheng J F, Rubin E M, Wood W G, Bowden D, Higgs D R (2006). A regulatory SNP causes a human genetic disease by creating a new transcriptional promoter. Science, 312(5777): 1215–1217
https://doi.org/10.1126/science.1126431
pmid: 16728641
|
| 5 |
Dehbozorgian J, Moghadam M, Daryanoush S, Haghpanah S, Imani Fard J, Aramesh A, Shahsavani A, Karimi M (2015). Distribution of alpha-thalassemia mutations in Iranian population. Hematology, 20(6): 359–362
https://doi.org/10.1179/1607845414Y.0000000227
pmid: 25553732
|
| 6 |
Derakhshan S M, Khaniani M S, Afkhami F, PourFeizi A H (2016). Molecular study of deletional and nondeletional mutations on the a-globin locus in the Azeri population of Northwestern Iran. Hemoglobin, 40(5): 319–322
https://doi.org/10.1080/03630269.2016.1240688
pmid: 27690152
|
| 7 |
Eftekhari H, Tamaddoni A, Mahmoudi Nesheli H, Vakili M, Sedaghat S, Banihashemi A, Azizi M, Youssefi Kamangar R, Akhavan-Niaki H (2017). A comprehensive molecular investigation of a-thalassemia in an Iranian cohort from different provinces of North Iran. Hemoglobin, 41(1): 32–37
https://doi.org/10.1080/03630269.2017.1299753
pmid: 28385057
|
| 8 |
Farashi S, Harteveld C L (2017). Molecular basis of a-thalassemia. Blood Cells Mol Dis
pmid: 29032940
|
| 9 |
Galanello R, Cao A (2011). Gene test review. Alpha-thalassemia. Genet Med, 13(2): 83–88
https://doi.org/10.1097/GIM.0b013e3181fcb468
pmid: 21381239
|
| 10 |
Harteveld C L, Higgs D R (2010). a-thalassaemia. Orphanet J Rare Dis, 5(1): 13
https://doi.org/10.1186/1750-1172-5-13
pmid: 20507641
|
| 11 |
HiggsD R, Gibbons R J ( 2010). The molecular basis of -thalassemia: a model for understanding human molecular genetics. Hematology/Oncology Clinics, 24(6): 1033–1054
|
| 12 |
Higgs D R, Wood W G (2008). Long-range regulation of a globin gene expression during erythropoiesis. Curr Opin Hematol, 15(3): 176–183
https://doi.org/10.1097/MOH.0b013e3282f734c4
pmid: 18391781
|
| 13 |
Ilan L, Osman F, Namer L S, Eliahu E, Cohen-Chalamish S, Ben-Asouli Y, Banai Y, Kaempfer R (2017). PKR activation and eIF2a phosphorylation mediate human globin mRNA splicing at spliceosome assembly. Cell Res, 27(5): 688–704
https://doi.org/10.1038/cr.2017.39
pmid: 28374749
|
| 14 |
Kanavakis E, Papassotiriou I, Karagiorga M, Vrettou C, Metaxotou-Mavrommati A, Stamoulakatou A, Kattamis C, Traeger-Synodinos J (2000). Phenotypic and molecular diversity of haemoglobin H disease: a Greek experience. Br J Haematol, 111(3): 915–923
pmid: 11122156
|
| 15 |
Keikhaei B, Slehi-Fard P, Shariati G, Khosravi A (2018). Genetics of Iranian Alpha-Thalassemia Patients: A Comprehensive Original Study. Biochem Genet,
https://doi.org/10.1007/s10528-018-9857-6
pmid: 29627922
|
| 16 |
Liu Y T, Old J M, Miles K, Fisher C A, Weatherall D J, Clegg J B (2000). Rapid detection of alpha-thalassaemia deletions and alpha-globin gene triplication by multiplex polymerase chain reactions. Br J Haematol, 108(2): 295–299
https://doi.org/10.1046/j.1365-2141.2000.01870.x
pmid: 10691858
|
| 17 |
Musallam K M ( 2013). Non-transfusion-dependent thalassemias. Haematologica, 98(6): 833–844
|
| 18 |
Onay H, Aykut A, Karaca E, Durmaz A, Solmaz A E, Çoğulu Ö, Aydınok Y, Vergin C, Özkınay F (2015). Molecular spectrum of a-globin gene mutations in the Aegean region of Turkey: first observation of three a-globin gene mutations in the Turkish population. Int J Hematol, 102(1): 1–6
https://doi.org/10.1007/s12185-015-1796-y
pmid: 25939702
|
| 19 |
Ribeiro D,Sonati M ( 2008). Regulation of human alpha-globin gene expression and alpha-thalassemia. Genet Mol Res, 7(4):1045–53
|
| 20 |
Sanger F, Nicklen S, Coulson A R (1977). DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA, 74(12): 5463–5467
https://doi.org/10.1073/pnas.74.12.5463
pmid: 271968
|
| 21 |
Satta S, Paglietti M E, Sollaino M C, Barella S, Moi P, Desogus M F, Demartis F R, Manunza L, Origa R (2017). Changes in HbA2 and HbF in alpha thalassemia carriers with KLF1 mutation. Blood Cells Mol Dis, 64: 30–32
https://doi.org/10.1016/j.bcmd.2017.03.007
pmid: 28342932
|
| 22 |
Singer S T (2009). Variable clinical phenotypes of a-thalassemia syndromes. Sci World J, 9: 615–625
https://doi.org/10.1100/tsw.2009.69
pmid: 19618088
|
| 23 |
Sollaino M C, Paglietti M E, Loi D, Congiu R, Podda R, Galanello R (2010). Homozygous deletion of the major alpha-globin regulatory element (MCS-R2) responsible for a severe case of hemoglobin H disease. Blood, 116(12): 2193–2194
https://doi.org/10.1182/blood-2010-04-281345
pmid: 20864588
|
| 24 |
Surapolchai P, Chuansumrit A, Sirachainan N, Kadegasem P, Leung K C, So C C (2017). A molecular study on the role of alpha-hemoglobin-stabilizing protein in hemoglobin H disease. Ann Hematol, 96(6): 1005–1014
https://doi.org/10.1007/s00277-017-2978-x
pmid: 28337528
|
| 25 |
Tamaddoni A, Hadavi V, Nejad N H, Khosh-Ain A, Siami R, Aghai-Meibodi J, Almadani N, Oberkanins C, Law H Y, Najmabadi H (2009). a-Thalassemia mutation analyses in Mazandaran province, North Iran. Hemoglobin, 33(2): 115–123
https://doi.org/10.1080/03630260902817297
pmid: 19373587
|
| 26 |
Valaei A, Karimipoor M, Kordafshari A, Zeinali S (2018). Molecular Basis of a-Thalassemia in Iran. Iran Biomed J, 22(1): 6–14
pmid: 29115104
|
| 27 |
Vernimmen D, Marques-Kranc F, Sharpe J A, Sloane-Stanley J A, Wood W G, Wallace H A, Smith A J, Higgs D R (2009). Chromosome looping at the human a-globin locus is mediated via the major upstream regulatory element (HS-40). Blood, 114(19): 4253–4260
https://doi.org/10.1182/blood-2009-03-213439
pmid: 19696202
|
| 28 |
Viprakasit V, Kidd A M, Ayyub H, Horsley S, Hughes J, Higgs D R (2003). De novo deletion within the telomeric region flanking the human a globin locus as a cause of a thalassaemia. Br J Haematol, 120(5): 867–875
https://doi.org/10.1046/j.1365-2141.2003.04197.x
pmid: 12614224
|
| 29 |
Wajcman H, Traeger-Synodinos J, Papassotiriou I, Giordano P C, Harteveld C L, Baudin-Creuza V, Old J (2008). Unstable and thalassemic a chain hemoglobin variants: a cause of Hb H disease and thalassemia intermedia. Hemoglobin, 32(4): 327–349
https://doi.org/10.1080/03630260802173833
pmid: 18654884
|
| 30 |
Wu M Y, He Y, Yan J M, Li D Z (2017). A novel selective deletion of the major a-globin regulatory element (MCS-R2) causing a-thalassaemia. Br J Haematol, 176(6): 984–986
https://doi.org/10.1111/bjh.14005
pmid: 26915575
|
| 31 |
Yu L H, Liu D, Cai R, Shang X, Zhang X H, Ma X X, Yan S H, Fang P, Zheng C G, Wei X F, Liu Y H, Zhou T B, Xu X M (2015). Changes in hematological parameters in a-thalassemia individuals co-inherited with erythroid Krüppel-like factor mutations. Clin Genet, 88(1): 56–61
https://doi.org/10.1111/cge.12443
pmid: 24930900
|
|
Viewed |
|
|
|
Full text
|
|
|
|
|
Abstract
|
|
|
|
|
Cited |
|
|
|
|
| |
Shared |
|
|
|
|
| |
Discussed |
|
|
|
|